
SOD1 Aggregation in Amyloid Lateral Sclerosis (ALS)
About ALS
Amyloid Lateral Sclerosis (ALS), also known as motor neuron disease (MND) or Lou Gehrig’s disease, is a neurodegenerative disorder that leads to a loss of motor neurons and death, often due to respiratory failure. Most cases are sporadic (sALS), but 5-10% are associated with a family history (fALS). This percentage may be higher due to patients’ limited knowledge of their family histories.1 Males are 1.2-1.5 times more likely than females to develop the disease,2 and old age is considered a risk factor with incident rates peaking between 60 and 75 years.3
SOD1 Structure and Function
Superoxide Dismutase 1 (SOD1) is a highly conserved, ubiquitously expressed enzyme that catalyzes the dismutation of superoxide radicals to protect organisms from oxidative damage.4 It is also known as Copper-Zinc Superoxide Dismutase, since it contains Cu and Zn centres involved in the reaction:4
SOD1-Cu2+ + O2•– → SOD1-Cu+ + O2
SOD1-Cu+ + O2•– + 2H+ → SOD1-Cu2+ + H2O2
O2•– + O2•– + 2H+ → O2 + H2O2
The oxygen (O2) and hydrogen peroxide (H2O2) products are both less toxic than superoxide radical (O2•–) reactant. The Cu active site is reduced from Cu2+ to Cu+ in the first step of the reaction, then oxidized back to Cu2+. H2O2 is subsequently converted into O2 and H2O by another enzyme.5 The Zn ion is linked to the Cu ion via a bridging imidazolate ligand6 and a secondary bridge where Asp 83 is hydrogen-bonded to both a Zn ligand and a Cu ligand.7 The Zn site is not redox active but serves to stabilize the SOD1 structure.8

Ribbon diagram showing Cu (orange) and Zn (grey) active sites in SOD1. Made in KiNG from PDB 2C9V. Source: Dcrjsr [CC BY 4.0 (https://creativecommons.org/licenses/by/4.0)]
Linking SOD1 and ALS
Approximately 20% of fALS cases are associated with SOD1 mutations.11 Over 150 mutations have been implicated.12 Most are point mutations;13 it is unclear whether all of these are sufficient to cause ALS as single mutations or if several could be needed in conjunction. SOD1 mutations can also occur in sALS,14 and have been associated with variations in fALS survival times.15 It has been suggested that mutations cause SOD1 to lose its ability to scavenge superoxide radicals, leading to increased oxidative stress, but it now seems more likely that SOD1 contributes to ALS by gaining a toxic function rather than losing its regular function.16 This toxic gain-of function is thought to be SOD1 misfolding and aggregation into oligomers and ultimately larger aggregates. This idea is supported by the presence of mutant SOD1–containing aggregates that increase in abundance as disease progresses.17
How SOD1 Mutations Lead to Aggregation
SOD1 usually undergoes the following post-translational modifications:
- Copper Insertion
- Zinc Insertion
- Dimerization
- Disulfide Bond Formation
Many fALS-associated mutations disrupt these post-translational modifications, preventing the proper structure and folding of the SOD1 protein.18 Misfolded SOD1 is prone to aggregation into soluble oligomers, via the formation of intermolecular disulfide bonds between the free cysteine residues of different SOD1 molecules and non-covalent interactions (hydrogen bonds) between beta strands of SOD1 subunits.16
Demetallation
Apo, or completely demetallated SOD1 is susceptible to oligomerization, as is zinc-deficient SOD1.16 FALS-associated mutations are thought to lower the zinc binding affinity of SOD1 by altering the zinc binding geometry.7 One possibility is that mutations that perturb the electrostatic loop allow solvent to access the metal sites, preventing metallation with Cu and Zn.19
Dimerization
Dimerization of SOD1 reduces the surface area that is accessible to solvent, which increases its stability. When dimerization is disrupted, unstable monomers are prone to aggregation. fALS-associated mutations occur on the dimer interphase and can cause dimers to dissociate into monomers which act as aggregation templates.18
Disulfide Bonding
Each SOD1 subunit contains a disulfide bond between two of cysteine residues (Cys 57 and Cys 146). Intrasubunit disulfide bonding increases SOD1 stability, so its interruption can result in aggregation of unstable species. Intrasubunit disulfide bond reduction is necessary for fibril initiation and promotes faster seeding of aggregates.20
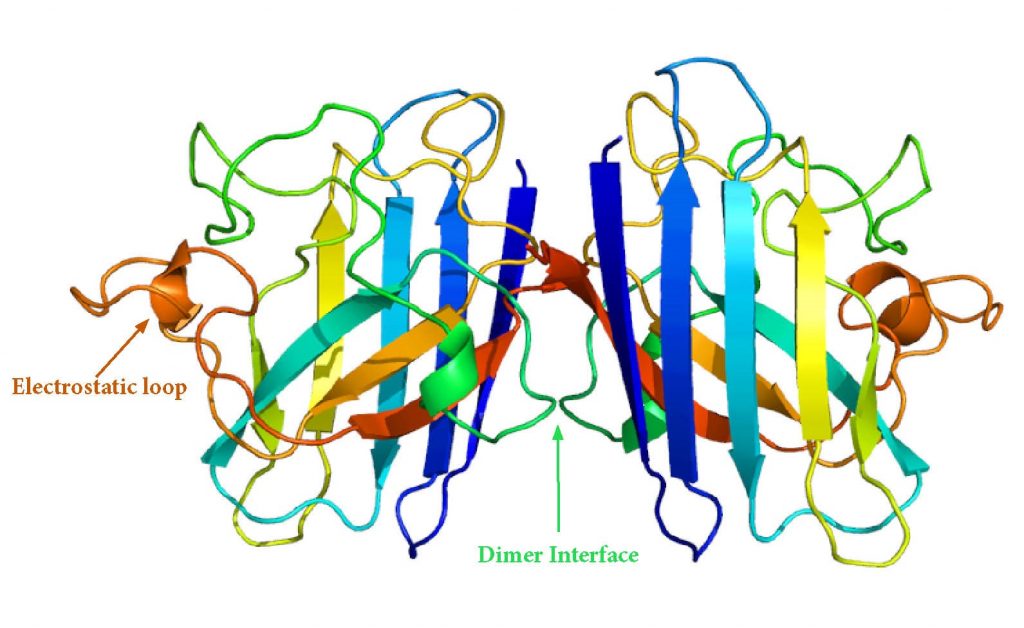
SOD1 Dimer Structure. Source: Adapted from Emw [CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)]
WT SOD1 Aggregation
WT SOD1 aggregation occurs in both fALS and sALS, and WT and mutant SOD1 are thought to aggregate via similar mechanisms.14 In vitro, WT and mutant SOD1 are both self-seeding and cross-seeding.21 Oxidation of WT SOD1 can produce similar effects as disease-causing mutations, and SOD1 fibrillation is heavily dependent on redox environment.22
Mechanisms of SOD1 Toxicity
As in other neurodegenerative diseases, the oligomeric form of SOD1 is likely more toxic than large SOD1 aggregates.23 Some hypothesize that aggregation is a protective mechanism against these toxic oligomers.24 These oligomers are thought to contain antiparallel, out-of-register β-sheet structures involving segment 28–38.25
There are several proposed mechanisms of toxicity for SOD1 oligomers:
Excitotoxicity
An increase in the extracellular glutamate concentration causes an influx of calcium into the postsynaptic neuron, which can result in mitochondrial damage and ultimately apoptosis.26 Riluzole, a drug that extends survival for ALS patients by 2-3 months,27 is thought to negate excitotoxicity.28
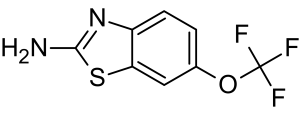
Riluzole Molecular Structure
Endoplasmic Reticulum Stress
Mutant SOD1 interacts with ER-associated degradation machinery (ERAD) and causes it to malfunction.29 This can also lead to apoptosis.
Axonal Transport Disruption
Mutant SOD1 can induce axonal transport defects by reducing microtubule stability and disrupting mitochondrial transport.30
Oxidative Stress and Mitochondrial Damage
Oxidative stress is associated with a variety of disease states, and oxidative stress is thought to contribute to the pathogenesis of sporadic ALS.31 The responsible ROS may be a product of mitochondrial dysfunction due to the accumulation of aggregated SOD1.32 The release of mitochondrial Ca2+ can also contribute to apoptosis.33
Non-Cell Autonomous Toxicity
ALS is considered to be non-cell autonomous, meaning that other non-neuronal cells, such as astrocytes and microglia also contribute to pathogenesis and disease progression.34
“Prion-Like” Cell-to-Cell Transmission
“Prion-like” refers to the ability of a protein to initiate self-propagating protein misfolding. While SOD1 is not infectious like PrPSc, and is unlikely to survive transmission between organisms, aggregated SOD1 can spread from cell to cell.35 Misfolded SOD1 can be released into the extracellular space when neurons die,35 or through exocytosis.36 It can then be taken up by other cells via fluid phase endocytosis.36
Interactions with Other Proteins
Alpha synuclein can cross-seed SOD137 and increases its oligomerization rate in mouse models.38 TDP-43 and FUS are also thought to trigger SOD1 misfolding.39
SOD1 PFFs
StressMarq recently launched SOD1 preformed fibrils (PFFs) for ALS research.

TEM of human SOD1 pre-formed fibrils (PFFs)
References
- Andersen, P.M. and A. Al-Chalabi, Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol, 2011. 7(11): p. 603-15.
- Manjaly, Z.R., K.M. Scott, K. Abhinav, L. Wijesekera, J. Ganesalingam, L.H. Goldstein, A. Janssen, A. Dougherty, E. Willey, B.R. Stanton, M.R. Turner, et al., The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotrophic Lateral Sclerosis, 2010. 11(5): p. 439-442.
- Chio, A., G. Logroscino, B.J. Traynor, J. Collins, J.C. Simeone, L.A. Goldstein, and L.A. White, Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology, 2013. 41(2): p. 118-130.
- Bergh, J. Structural investigation of SOD1 aggregates in ALS : identification of prion strains using anti-peptide antibodies. 2018.
- Bosco, D. A. (2015) The Role of SOD1 in Amyotrophic Lateral Sclerosis. Nature Education 8(3):4
- Bertini, Ivano and Gray, Harry B. and Lippard, Stephen J. and Valentine, Joan Selverstone (1994) Bioinorganic Chemistry. University Science Books , Mill Valley, CA. 5. Dioxygen Reactions
- Rakhit, R., Chakrabartty, A. (2006). Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim et Biophys Acta. 1762: 1025-1037.
- Messerschmidt, A. Comprehensive Natural Products II: Chemistry and Biology. 8.14 Copper Metalloenzymes. p. 489-545.
- Valentine, J. S., Doucette, P. A., and Zittin Potter, S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Rev. Biochem. 74, 563–593
- Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW. 1995. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci USA 92:954–8.
- Rosen, D.R., Siddique, T., Patterson, D. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362 (1993), p. 59-62
- Brasil, A.A., Magalhaes, R.S.S., De Carvalho, M.D.C., Paival, I., Gerhardt, E., Pereira, M.D., Outeiro, T.F., Eleutherio, E.C.A. Implications of fALS Mutations on Sod1 Function and Oligomerization in Cell Models. Mol Neurobiol (2018) 55:5269-5281.
- Guegan C, Przedborski S. Programmed cell death in amyotrophic lateral sclerosis (2003) J Clin Invest 111:153–161.
- Gruzman A, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2007; 104:12524–12529.
- Alemasov, N.A., Ivanisenko, N.V., Medvedev, S.P., Zakian, S.M, Kolchanov, N.A., Ivanisenko, V.A. Dynamic properties of SOD1 mutants can predict survival time of patients carrying familial amyotrophic lateral sclerosis. J Biomol Struct Dyn. (2017). 35(3):645-656.
- Banci L, Bertini I, Boca M, Girotto S, Martinelli M, et al (2008) SOD1 and Amyotrophic Lateral Sclerosis: Mutations and Oligomerization. PLoS ONE 3(2):e1677.
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, et al. (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281: 1851–1854.
- McAlary, L., Aquilina, J.A., Yerbury, J.J. (2016). Susceptibility of Mutant SOD1 to Form a Destabilized Monomer Predicts Cellular Aggregation and Toxicity but Not In vitro Aggregation Propensity. Front Neurosci. 10:499.
- Schmitt, N.D., Agar, J.N. Parsing Disease-relevant Protein Modifications from Epiphenoma: Perspectives on the Structural Basis of SOD1-Mediated ALS. (2017). J Mass Spectrom. 52(7): 480–491
- Chattopadhyay, M., Nwadibia, E., Strong, C.D., Gralla, E.B., Valentine, J.S., Whitelegge, J.P. The Disulfide Bond, but Not Zinc or Dimerization, Controls Initiation and Seeded Growth in Amyotrophic Lateral Sclerosis-linked Cu,Zn Superoxide Dismutase (SOD1) Fibrillation. J Biol Chem. (2015) 290(51):30624-36.
- Bunton-Stasyshyn, R.K.A., Saccon, R.A., Fratta, P., Fisher, E.M.C. SOD1 function and its implications for ALS. The Neuroscientist 2015, Vol. 21(5) 519–529
- Alvarez-Zaldiernas, C., Lu, J., Zheng, Y., Yang, H., Blasi, J., Solsona, C., Holmgren, A. Cellular Redox Systems Impact the Aggregation of Cu,Zn Superoxide Dismutase Linked to Familial Amyotrophic Lateral Sclerosis. J Biol Chem. (2016). 291(33):17197-17208.
- Ross CA, Poirier MA (2006) Protein Aggregation and Neurodegenerative Disease. Nat Med 10: S10–17.
- Zhu, C., Beck, M.V., Griffith, J.D., Deshmukh, M., Dokholyan, N.V. (2018). Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 115(18):4661-4665.
- Sangwan, S. et al. (2017). Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). PNAS 114(33):8770-8775.
- Van Den Bosch, L., Van Damme, P., Bogaert, E., Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. BBA – Molecular Basis of Disease. 1762(11-12):1068-1082.
- Miller, R., Mitchell, J., Moore, D. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012; 3
- Bensimon, G., L. Lacomblez, and V. Meininger, A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med, 1994. 330(9): p. 585-91.
- Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., Noguchi, T., Matsuzawa, A., Takeda, K., Ichijo, H. (2008). ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes & Dev. 22:1451-1464.
- De Vos, K.J., Hafezparast, M. (2017). Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? 105: 283-299.
- D’Amico E., Factor-Litvak P., Santella R. M., Mitsumoto H. (2013). Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 65, 509–527.
- Ferri A., Cozzolino M., Crosio C., Nencini M., Casciati A., Gralla E. B., et al. . (2006). Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Natl. Acad. Sci. USA 103, 13860–13865
- Chinnery PF, Elliott HR, Hudson G, et al. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol. 2012;41(1):177–187.
- Ilieva, H., Polymenidou, M., Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. (2009) J Cell Biol. 187(6):761-772.
- Grad, L.I., Cashman, N.R. (2014). Prion-like activity of Cu/Zn superoxide dismutase, Prion, 8:1, 33-41.
- Lee, S., Kim, H. (2015) Prion-like Mechanism in Amyotrophic Lateral Sclerosis: are Protein Aggregates the Key? Exptl Neurobiol. 24(1):1-7.
- Helferich, A.M, McLean, P.J., Weishaupt, J.H., Danzer, K.M. (2016), Alpha-synuclein interacts with SOD1 and promotes its oligomerization. J Neurol & Neuromed. 1(7):28-30.
- Koch, Y., Helferich, A.M., Steinacker, P., Oeckl, P., Walther, P., Weishaupt, J.H., Danzer, K.M., Otto, M. Aggregated a-Synuclein Increases SOD1 Oligomerization in a Mouse Model of Amyotrophic Lateral Sclerosis (2016). 186(8):2152-2161.
- Pokrishevsky, E., L.I. Grad, and N.R. Cashman, TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Scientific Reports, 2016. 6: p. 22155.
Leave a Reply