
A53T Alpha Synuclein in Parkinson’s Disease
Alpha Synuclein in Parkinson’s Disease
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the aggregation of alpha synuclein into Lewy bodies in the brain. Alpha synuclein is a 14-kDa protein encoded by the SNCA gene. There is conflicting evidence regarding the native state of alpha synuclein: it may be an unfolded monomer,1 folded tetramer, or a dynamic equilibrium between these and other oligomers.2 In Parkinson’s Disease, these smaller forms aggregate into protofibrils, fibrils, and ultimate Lewy bodies, which are thought to cause neuronal dysfunction and death. There is also the possibility that the oligomers and protofibrils are the neurotoxic species3-6 while Lewy bodies are neuroprotective.7
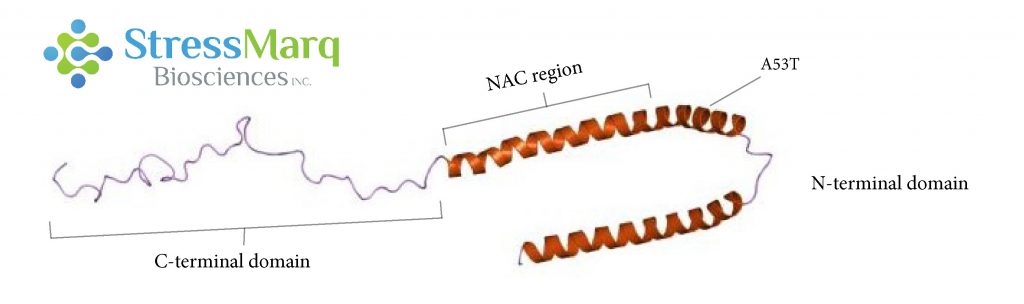
Alpha synuclein consists of three main regions: the C-terminal domain, the NAC region, and the N-terminal domain. The A53T mutation is found in the N-terminal domain.
What is the A53T mutation?
A53T is a missense point mutation, meaning one amino acid is changed: the 53rd amino acid is changed from alanine to threonine. This mutation is caused by guanine being changed to adenine at position 209 of the SNCA gene (G209A).8
What is the role of the A53T mutation in Parkinson’s Disease?
The A53T mutation has been linked to autosomal dominant, early-onset PD, first documented in families of Italian and Greek descent.8 It has also been documented in a Korean family.9 A53T mutation results in an earlier age of onset and a shorter disease duration.8 Even though most cases of PD are sporadic, not inherited, and do not involve the A53T mutation, understanding the role of A53T could help researchers better understand mechanisms of alpha synuclein aggregation and develop better models and therapies.
A53T Aggregation
A53T alpha synuclein fibrillizes in solution faster than wild-type (WT) alpha synuclein.10 The mutation, however, has a greater effect on the rate of alpha synuclein protofibril formation than on the rate of fibril elongation.11 The fact that A53T and A30P mutations are both linked to early-onset PD and promote protofibril formation suggests that protofibrils contribute to the neurodegeneration seen in PD.12
Why does A53T alpha synuclein fibrillize faster than WT alpha synuclein?
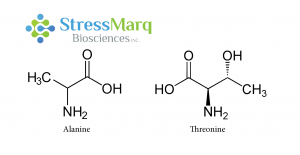
Alanine and Threonine have similar structures, but substituting Threonine for Alanine in alpha synuclein has significant effects on alpha synuclein fibrillization.
The A53T mutation only involves a single changed amino acid, and alanine and threonine are structurally similar, so why is there such a significant effect? A53T and other disease-causing mutations occur in the N-terminal region of the alpha synuclein protein. Theoretical models indicate that the A53T mutation causes long-range interactions between the NAC region and the N- or C-terminal regions of alpha synuclein to disappear, leading to an increase in beta-sheet formation.13 NMR measurements indicate that the A53T mutation could extend and stabilize beta-sheet structures involved in oligomerization and fibrillization.14
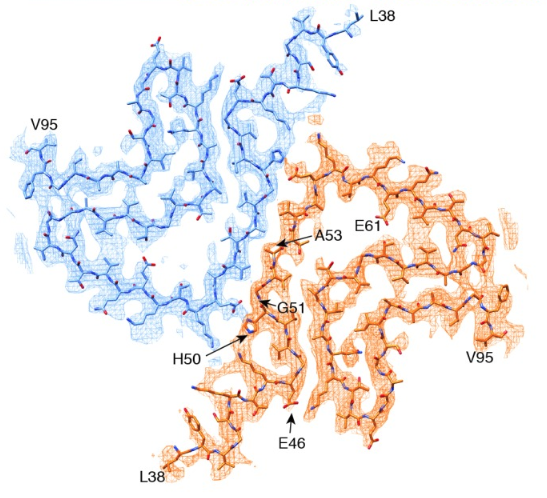
The cryo-EM-determined structure of alpha synuclein shows most disease-causing mutations to be located at the interface between two protofilaments.15 Source: doi.org/10.7554/eLife.36402.001 © 2018, Guerrero-Ferreira et al.
Cryo-EM analysis revealed that alpha synuclein fibrils consist of two protofilaments twisted together into left-handed helices.16 The A53T mutation, along with four of the five other early-onset PD mutations, is located at the interphase between the two protofilaments.16 Introducing a hydrophilic residue such as threonine disrupts this “steric-zipper interphase” and changes the structure of the fibrils.16
Effects of the A53T Mutation
Regardless of the mechanism of A53T alpha synuclein aggregation, it has devastating downstream effects. A53T alpha synuclein expression in mice leads to motor deficits, paralysis, and death.17 A53T alpha synuclein has a greater tendency than WT alpha synuclein to form annular and tubular protofibrils, and A53T alpha synuclein protofibrils bind tightly to phospholipid vesicles, leading to their permeabilization.12 These lipid vesicles can induce fibril production by enhancing nucleation of monomers.18 The A53T mutation is thought to promote fibrillization by either changing the structure of vesicle pores or by promoting the formation permeabilizing protofibrils over other species.19
Dopamine
Dopamine is a neurotransmitter that regulates speech and movement. Parkinson’s Disease motor deficits are caused by a loss in dopamine-secreting (dopaminergenic) neurons. Mice overexpressing A53T alpha synuclein experience impaired dopamine signaling prior to neurodegeneration,20 and rats expressing A53T alpha synuclein showed greater losses of dopaminergic neurons than their counterparts expressing WT alpha synuclein.21 A53T alpha synuclein expression is also thought to make cells more vulnerable to dopamine toxicity.22
Synapses
Synapses are components of neurons responsible for the transfer of information between neurons via synaptic vesicles.23 Both A53T and WT alpha synuclein suppress presynaptic transmission by depleting the synaptic vesicle recycling pool.24 A53T alpha synuclein expression also produces postsynaptic deficits.25 These postsynaptic deficits are caused by the hyperphosphorylation of tau and its mislocalization to dendritic spines.24
Microglia
Microglia are responsible for synaptic pruning and respond to injuries by producing inflammatory mediators.26 Activated microglia change morphologies, produce reactive oxygen species (ROS), and release inflammatory cytokines.27 In Parkinson’s Disease, they may become overactivated and contribute to the depletion of dopaminergic neurons.27 A53T alpha synuclein activates microglia more strongly than WT or other mutants, leading to microgliosis and a subsequent inflammatory state.27 A53T alpha synuclein expression also makes cells more vulnerable to oxidative stress.28
Astrocytes
When A53T alpha synuclein is selectively expressed in mouse astrocytes, it leads to rapid paralysis.29 This is thought to be due decreased glutamate transporter expression disruption leading to downstream activation of microglia.29
Mitochondrial Dysfunction and Endoplasmic Reticulum Stress
A53T alpha synuclein-induced inflammation and oxidative stress affect both the mitochondria and the endoplasmic reticulum (ER). A53T expression causes mitochondrial dysfunction involving impaired mitochondrial membrane potential and respiratory function.30 This in turn leads to mitochondrial autophagy.31 Overactivation of autophagy may lead to the mitochondrial loss and resultant neurodegeneration.31 However, it may also have some protective effects by clearing aggregated proteins.31 A53T neurons showed impaired mitochondrial motility that could be reversed by rapamycin, an autophagy inducer.30
In differentiated PC12 cells, increasing A53T alpha synuclein expression led to up to 40% cell death due to increased intracellular ROS level and decreased proteasome activity.32 In this case, ER stress and mitochondrial function involved the release of mitochondrial cytochrome C and elevated caspase-3, -9, and -12 activities, and led to cell death.32
Therapeutic Approaches
Preventing the neurodegeneration associated with A53T mutation could lead to potential PD therapies. A COX-1 inhibitor extends the lifespan of A53T alpha synuclein-expressing mice,29 and Cyclosporin A, Z-Vad, and Caspase-3 and -9 inhibitors reduced cell death from ER and mitochondrial stress.32 Although the A53T mutation is not present in the majority of PD cases, WT alpha synuclein may have similar but slower mechanisms of fibrillization and subsequent neurodegeneration. A53T mutated alpha synuclein was more effective than WT alpha synuclein in establishing a PD model in rats, and resulted in a model representative of early-onset PD.21 An enhanced understanding of A53T alpha synuclein fibrillization and inhibition mechanisms could help those with the A53T mutation in addition to other PD patients.
StressMarq recently launched two A53T mutant alpha synuclein proteins for neurodegenerative disease research: monomers and pre-formed fibrils.
References
- Fauvet B, et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–64.
- Dehay B, Bourdenx M, Gorry P, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14(8):855-866.
- Karpinar DP, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009;28:3256–68.
- Winner B, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–9.
- Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. 2012;149:1048–59.
- Danzer KM, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–32.
- Tanaka M, et al. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–31.
- Polymeropoulos, M. H. Mutation in the -Synuclein Gene Identified in Families with Parkinson’s Disease. Science, 1998;276(5321), 2045–2047. doi:10.1126/science.276.5321.2045
- Ki C.S. Stavrou E.F. Davanos N. Lee W.Y. Chung E.J. Kim J.Y. Athanassiadou A. The Ala53Thr mutation in the alpha-synuclein gene in a Korean family with Parkinson disease. Clin Genet. 2007 May;71(5):471-3.
- Conway, K.E., Harper, J.D., & Lansbury, P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. 1998, 4(11):1318-20
- Flagmeier, P. et al. (2016). Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. PNAS. 113(37):10328-10333.
- Lashuel, H.A., Petre, B.M, Wall, J. et al. α-Synuclein, Especially the Parkinson’s Disease-associated Mutants, Forms Pore-like Annular and Tubular Protofibrils. J Mol Biol. 2002 Oct 4;322(5):1089-102
- Coskuner, O., Wise-Scira, O. Structures and Free Energy Landscapes of the A53T Mutant-Type α‑Synuclein Protein and Impact of A53T Mutation on the Structures of the Wild-Type α‑Synuclein Protein with Dynamics. ACS Chem. Neurosci. 2013, 4, 1101−
- Russel, R., Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J Biol Chem. 2001 Dec 7;276(49):45996-6003.
- Guerrero-Ferreira, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H. Cryo-EM structure of alpha-synuclein fibrils. Elife. 2018 Jul 3;7. pii: e36402
- Li, Y. et al. Amyloid fibril structure of α-synuclein determined by cryoelectron microscopy. Cell Research (2018) 0:1–7
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ et al. (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34: 521-533.
- Galvagnion C, et al. (2015) Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol 11(3):229–234.
- Volles, M., Lansbury, P.T. Zeroing in on the Pathogenic Form of α-Synuclein and Its Mechanism of Neurotoxicity in Parkinson’s Disease. Biochem. 2003, 42(26). 10.1021/bi030086j
- Kurz A, Double KL, Lastres-Becker I, Tozzi A, Tantucci M, et al. (2010) A53T-Alpha-Synuclein Overexpression Impairs Dopamine Signaling and Striatal Synaptic Plasticity in Old Mice. PLoS ONE 5(7): e11464.
- Lu, J., Sun, F. et al. (2015). Comparison between alpha synuclein wild-type and A53T mutation in a progressive PD model. Biochem & Biophys Res. Commun. 464(4):988-993.
- Tabrizi, S. J.; Orth, M.; Wilkinson, J. M.; Taanman, J. W.; Warner, T. T.; Cooper, J. M.; Schapira, A. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Mol. Genet. 2000, 9, 2683-2689
- Lepeta K, Lourenco MV, Schweitzer BC, et al. Synaptopathies: synaptic dysfunction in neurological disorders – A review from students to students. J Neurochem. 2016;138(6):785-805.
- Nemani VM, Lu W, Berge V, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. 2010;65(1):66-79.
- Teravskis, P.J. et al. (2018). A53T mutant alpha-synuclein induces tau dependent postsynaptic impairment independent of neurodegenerative changes. Neurosci; 10.1523
- Joe EH, Choi DJ, An J, Eun JH, Jou I, Park S. Astrocytes, Microglia, and Parkinson’s Disease. Exp Neurobiol. 2018;27(2):77-87.
- Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, et al. (2016) Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 11(9): e0162717
- Kanda, S.; Bishop, J. F.; Eglitis, M. A.; Yang, Y.; Mouradian, M. M. Enhanced vulnerability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience 2000, 97, 279-284.
- Gu, X., Long, C. et al. (2010). Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Molecular Brain. 3:12.
- Li L, Nadanaciva S, Berger Z, Shen W, Paumier K, et al. (2013) Human A53T α-Synuclein Causes Reversible Deficits in Mitochondrial Function and Dynamics in Primary Mouse Cortical Neurons. PLoS ONE 8(12): e85815
- Choubey V, Safiulina D, Vaarmann A, et al. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem. 2011;286(12):10814-24.
- Smith, W.W. et al. (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Gen. 14(24):3801-3811.
Leave a Reply