
Tau Mutations and their Association with Neurodegenerative Disease
The tubulin-associated unit (tau) is a microtubule-binding phosphoprotein capable of promoting the assembly and stability of microtubules. There are six different isoforms of tau in normal human brain produced from alternative splicing of a single gene, ranging from 352 to 441 amino acids in length. At the carboxy-terminal end of tau lies the microtubule-binding region. Half of the six isoforms contain three repeats of this microtubule-binding region (3R tau), whereas the other half contain four repeats of this region (4R tau). This is due to the exclusion or inclusion of exon 10 by alternative splicing. In normal human brain the ratio of 3R tau to 4R tau is expected to be approximately equal (1, 2).
Tau is normally found in the axons of neurons but in tauopathies, the normally soluble tau, forms insoluble fibrils in cell bodies and dendrites. These hyper-phosphorylated tau lesions or plaques are a characteristic of neurodegenerative diseases such as Alzheimer’s disease and a disorder known as inherited frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). In Alzheimer’s disease and FTDP-17 the neurofibrillary tangles can be observed as consisting of paired helical filaments (PHFs) with the repeat region forming the core of the filament (2).
What causes tau to form filaments?
It was initially unknown whether tau accumulation was a symptom or cause of neurodegenerative disease. This was not answered until researchers discovered a link between a mutation on the tau gene on chromosome 17 and frontotemporal dementia (3). A considerable number of tau mutations have now been discovered in Alzheimer’s disease and FTDP-17 patients. Mutations have been identified within coding regions (missense and deletion mutations) and non-coding regions (at the splice-donor site near exon 10). Mutations can be isoform specific, those found in exons 9, 12 and 13 affect all six tau isoforms, whereas mutations in exon 10 (examples include N279K, ΔK280, P301L, P301S and S305N) only affect the 4R tau isoform.
It is proposed that many of the known tau mutations cause their deleterious effects by either reducing the ability of tau to bind microtubules or encouraging fibril formation. The mechanism of which is not fully resolved but could be due to increasing the ratio of the 4R tau versus 3R tau, or by changing the level of overall tau expression (4, 5). Beta-sheet formation in the microtubule-binding region, around exons 10 and 11 is a proposed mechanism by which tau polymerizes into paired helical filaments (PHF).
Specific mutations ΔK280 and P301L found in 4R tau have been shown to increase the tendency of tau to aggregate. Both these mutations confer properties favorable to β-sheet formation and are associated with frontotemporal dementia. Many of the mutations around exon 10 are intronic and alter the ratio of the 3R tau and 4R tau isoforms towards an excess of the 4R isoform, the exception being ΔK280 where the 3R form predominates (5, 6).
Supporting the study of tau aggregation
StressMarq are the first to offer Tau preformed fibrils (PFFs) and filaments for neuroscience research. Tau PFFs induce endogenous tau phosphorylation and aggregation, leading to disease pathology in vivo. Choose from our expanding portfolio of wild-type or mutated tau isoforms.
Mutations include a C322A mutated dGAE fragment, which lacks cysteine residues, and assembles into PHF-like fibrils in vitro in the absence of additives or templates (catalog# SPR-462) (7). The P301L mutation in the K18 truncated form (catalog# SPR-330). The P301S mutation in the full length 2N4R of human tau (catalog# SPR-463) or mouse tau (catalog# SPR-475), and the ΔK280 deletion mutation in K18 truncated tau (catalog# SPR-477).
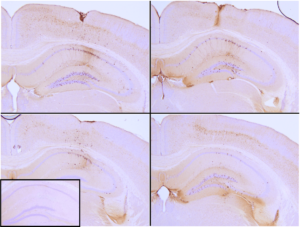
Immunohistochemical analysis of P301L mouse hippocampus injected with K18 P301L tau PFFs (SPR-330) shows seeding of tau pathology at injection site nine weeks post-injection. AT8 (pSer202/pThr205) tau antibody shows tangle-like inclusions. Inset: negative control. Experiments performed at reMYND N.V.
REFERENCES
- Goedert, M. and Jakes, R. EMBO J., (1990) 9:4225-4230
- Goedert, M. and Jakes, R. Biochim Biophys Acta., 1739 (2005) 240–250
- Wilhelmsen, K.C. et al. Am. J. Hum. Genet., 55 (1994) 1159-1165
- Goedert, M. et al. Biochim Biophys Acta., (2000) 1502(1):110-21
- Von Bergen, M. et al. JBC, (2001) 276(51):48165-48174.
- Alberici, A. et al. J Neurol Neurosurg Psychiatry, (2004) 75:1607-1610.
- Al-Hilaly, Y.K. et al. J. Mol. Biol., (2017) 429(23):3650-3665
Leave a Reply