
Wild-Type Tau in Alzheimer’s Disease
The microtubule-associated protein tau, encoded by the MAPT gene, is expressed predominantly in neurons. Here, it exists as six different monomeric isoforms ranging from 352-441 amino acids in length1. Under normal conditions, tau monomers perform an essential role in stabilizing neuronal microtubules and regulating axonal transport. However, under pathological conditions, detachment of tau monomers from neuronal microtubules results in the formation of intracellular aggregates, leading to neuronal cell death. To date, over 20 different tauopathies have been described, with Alzheimer’s disease being the most prevalent. Of these, around 10% are due to dominant mutations in the MAPT gene and are collectively termed frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), although it has recently been proposed that this name be retired2,3. The remainder occur sporadically and involve only the wild-type tau protein2.
How is wild-type tau involved in sporadic neurodegenerative disease?
Under pathological conditions, wild-type tau monomers are hyperphosphorylated and become detached from negatively-charged neuronal microtubules. They then undergo misfolding and conformational change, accumulating in the cytoplasm to trigger the fibrillogenesis pathway – the spontaneous formation of oligomers that can fibrillize into intracellular paired helical filaments (PHFs) and neurofibrillary tangles (NFTs)4. Tau oligomers, PHFs and NFTs can all be secreted and transmitted to neighboring cells, where their uptake seeds further oligomerization or aggregation, contributing to disease progression. Although NFTs have long been considered the main cause of cognitive impairment in patients with tauopathies (e.g., NFT numbers have been correlated to the degree of dementia in Alzheimer’s disease), it has more recently been suggested that tau oligomers contain the principal toxic tau species4,5.
How do mutant tau fibrils affect wild-type tau monomers?
Pathogenic mutations in the MAPT gene usually affect the tau coding sequence, leading to single amino acid changes that alter the protein’s primary structure. Depending on exactly where they occur, such mutations can augment tau aggregation. Studies aimed at understanding the role of specific tau mutations in disease pathogenesis include a comparison of fibril formation by various recombinant tau mutants, where both the G272V and P301L mutations were shown to increase the efficiency of filament extension6. Additionally, using P301L/V337M mutant fibrils to seed the aggregation of wild-type tau monomers has revealed these to produce a novel fibrillar conformation that is maintained over multiple seeding reactions2.
What is the role of wild-type tau in disease models?
Wild-type tau provides insights into tauopathies such as Alzheimer’s disease in a myriad of different ways. Popular strategies include recombinantly over-expressing tau to determine its impact on normal physiological processes and comparing the seeding capacity of different tau isoforms to discover how these are involved in the onset and progression of disease. With the majority of tauopathies being sporadic, learning more about wild-type tau is vital to develop effective therapies.
Supporting the study of tauopathies
StressMarq offers a wide selection of reagents for studying tau and its role in neurodegenerative disease. These include our Tau441 (2N4R) Wild-Type Monomer (catalog# SPR-479) that can be combined with our Tau441 (2N4R) Wild-Type Preformed Fibrils (catalog# SPR-480) for studying tau aggregation in vitro. We also sell Wild-Type Truncated Tau Fragment (AA297-391) (dGAE) Preformed Fibrils (catalog# SPR-461) and accompanying Truncated Tau Fragment (AA297-391) (dGAE) monomer (catalog# SPR-444) for aggregation studies. For more information on Tau including a list of all our Tau fibrilized proteins and a short video on Tau PFFs, check out our webpage dedicated to Tau.
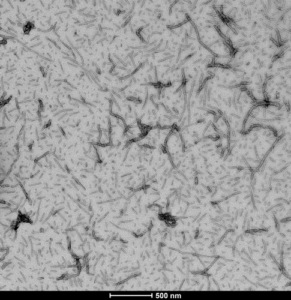
TEM of human recombinant Tau441 (2N4R), Wild-Type Preformed Fibrils (catalog# SPR-480)
Leave a Reply